人工智能(AI)一直在迅速发展,但对人类来说,强大的模型却是个「黑匣子」。
我们不了解模型内部的运作原理,不清楚它得出结论的过程。
然而最近,波恩大学(University of Bonn)的化学信息学专家Jürgen Bajorath教授和他的团队取得了重大突破。
他们设计了一种技术,揭示了药物研究中使用的某些人工智能系统的运行机制。
他们的研究结果表明,这些人工智能模型主要依赖于回忆现有数据,而不是学习特定的化学相互作用,来预测药物的有效性。
——也就是说,AI预测纯靠拼凑记忆,机器学习实际上并没有学习!
他们的研究结果最近发表在《自然机器智能》(Nature Machine Intelligence)杂志上。

论文地址:https://www.nature.com/articles/s42256-023-00756-9
在医药领域,研究人员正在狂热地寻找有效的活性物质来对抗疾病——哪种药物分子最有效?
通常,这些有效的分子(化合物)会对接在蛋白质上,蛋白质作为触发特定生理作用链的酶或受体。
在特殊情况下,某些分子还负责阻断体内的不良反应,例如过度的炎症反应。
可能的化合物数量巨大,寻找有效的化合物就像大海捞针一样。
因此,研究人员首先使用AI模型来预测,哪些分子最能与各自的靶蛋白对接并牢固结合。然后在实验研究中,更详细地进一步筛选这些候选药物。
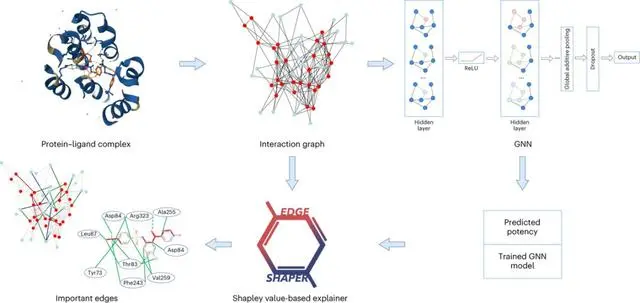
自人工智能发展以来,药物发现研究也越来越多地采用AI相关的技术。
比如图神经网络(GNN),适用于预测某种分子与靶蛋白结合的强度。
图由表示对象的节点和表示节点之间关系的边组成。在蛋白质与配体复合物的图表示中,图的边连接蛋白质或配体节点,表示物质的结构,或者蛋白质和配体之间的相互作用。
GNN模型使用从X射线结构中提取的蛋白质配体相互作用图,来预测配体亲和力。
Jürgen Bajorath教授表示,GNN模型对于我们来说就像一个黑匣子,我们无法得知它如何得出自己的预测。

Jürgen Bajorath教授任职于波恩大学LIMES研究所、波恩-亚琛国际信息技术中心(Bonn-Aachen International Center for Information Technology)和拉玛机器学习与人工智能研究所(Lamarr Institute for Machine Learning and Artificial Intelligence)。
人工智能如何工作?
来自波恩大学化学信息学的研究人员,与罗马Sapienza大学的同事一起,详细分析了图神经网络是否真的学习到了蛋白质与配体的相互作用。
研究人员使用他们专门开发的「EdgeSHAPer」方法分析了总共六种不同的GNN架构。
EdgeSHAPer程序可以判断GNN是否学习了化合物和蛋白质之间最重要的相互作用,或者是通过其他的方式来得出预测。
科学家们使用从蛋白质配体复合物结构中提取的图训练了六个GNN,——化合物的作用方式以及与靶蛋白的结合强度已知。
然后,在其他复合物上测试经过训练的GNN,并使用EdgeSHAPer分析GNN如何产生预测。
「如果GNN按照预期行事,它们需要学习化合物和靶蛋白之间的相互作用,并且通过优先考虑特定的相互作用来给出预测」。
然而,根据研究小组的分析,六个GNN基本上都没有做到这一点。大多数GNN只学会了一些蛋白质与药物的相互作用,主要集中在配体上。
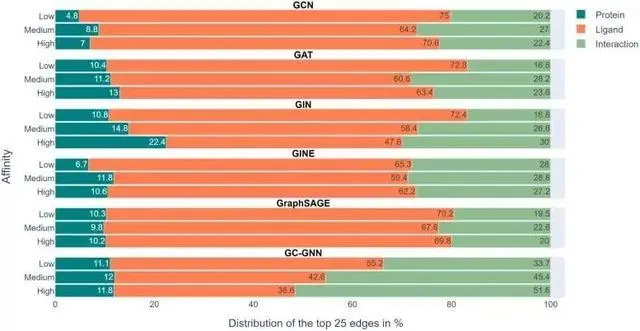
上图展示了在6个GNN中的实验结果,色标条表示用EdgeSHAPer确定的每个预测的前25个边中蛋白质、配体和相互作用所占的平均比例。
我们可以看到,代表绿色的相互作用本该是模型需要学到的,然而在整个实验中所占的比例都不高,而代表配体的橙色条占了最大的比例。
为了预测分子与靶蛋白的结合强度,模型主要「记住」了它们在训练过程中遇到的化学相似分子及其结合数据,而不管靶蛋白如何。这些被记住的化学相似性基本上决定了预测。
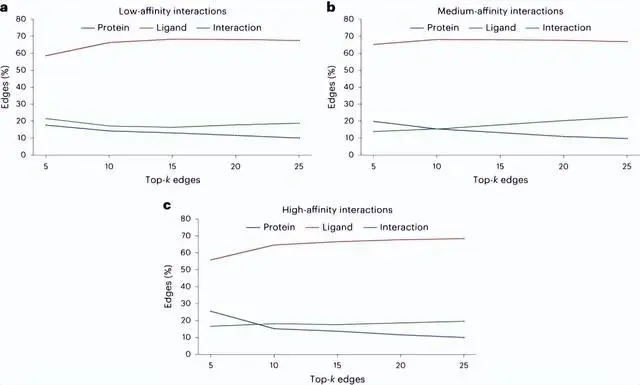
这让人想起「聪明的汉斯效应」(Clever Hans effect),——就像那匹看起来会数数的马,实际上是根据同伴面部表情和手势的细微差别,来推断出预期的结果。
这或许意味着,GNN所谓的「学习能力」可能是站不住脚的,模型的预测在很大程度上被高估了,因为可以使用化学知识和更简单的方法进行同等质量的预测。
不过,研究中也发现了另外一个现象:当测试化合物的效力增加时,模型倾向于学习到更多的相互作用。
也许通过修改表征和训练技术,这些GNN还能朝着理想的方向进一步改进。不过,对于可以根据分子图学习物理量的假设,一般来说应该谨慎对待。
「人工智能不是黑魔法。」









暂无评论内容